- بروفايل ميدار
- وثائقيات ميدار
- ميدار ChatGPT
- من العالم
- مقالات ميدار
- تقارير ميدار
- صحة
- كريبتو
- تكنولوجيا وسيارات
- مال وأعمال
- كتب ميدار
ميدار.نت - دبي
مرض التلاسيميا، أو الثلاسيميا، هو اضطراب وراثي في الدم، ينتقل عبر الجينات من الآباء إلى الأطفال، ويحدث عندما لا ينتج الجسم كمية كافية من بروتين الهيموغلوبين، الموجود في خلايا الدم الحمراء، ما يؤدي إلى عدم عمل الكريات الحمر بشكل صحيح فتموت بسرعة، لذا يكون عدد خلايا الدم السليمة التي تنتقل في مجرى الدم أقل منها لدى الإنسان الطبيعي.
يتكون الهيموغلوبين من نوعين من البروتينات، ألفا غلوبين، وبيتا غلوبين، ويحدث مرض التلاسيميا عندما يكون هناك خلل في جين يساعد في التحكم في إنتاج أحد هذه البروتينات. لذلك صنفت التلاسيميا إلى نوعين أساسيين:
_ تلاسيميا ألفا، حيث تكون الجينات المرتبطة ببروتين ألفا غلوبين مفقودة أو متغيرة، ويصاب بهذا النوع الأشخاص في مناطق جنوب شرق آسيا، والشرق الأوسط، والصين، وافريقيا.
_ تلاسيميا بيتا، وتحدث عندما تؤثر عيوب جينية متماثلة على إنتاج بروتين بيتا غلوبين، وتحدث غالبا عند الآسيويين، والأمريكيين من أصل إفريقي.
ويتضمن كلا النوعين التلاسيميا الكبرى والتلاسيميا الصغرى، فالتلاسيميا الكبرى تحدث إذا ورث الطفل الخلل الجيني من الوالدين معا، أما مرض التلاسيميا الصغرى فيحدث إذا تلقى الطفل الخلل الجيني من أحد الوالدين فقط.
تنشأ مؤشرات مرض التلاسيميا لدى بعض الأطفال عند الولادة، وتظهر لدى أطفال آخرين خلال العامين الأوليين من العمر، وبعض الأطفال المصابون بالتلاسيميا الصغرى لا يواجهون أعراض مرض التلاسيميا.
وتشمل أعراض هذا المرض:
_ الإرهاق.
_ ضعف الجسم.
_ تشوهات في عظام الوجه.
_ شحوب الجلد واصفراره.
_ اللون الغامق للبول.
_ انتفاخ البطن.
_ بطء النمو.
في حال اشتباه الطبيب بإصابة الطفل بمرض التلاسيميا، يطلب إجراء فحوصات الدم، للكشف عن عدد خلايا الدم الحمراء، وحجمها ولونها وشكلها، كما يمكن من خلال هذه الفحوصات تحليل الحمض النووي للكشف عن الجينات الطافرة.
كما يمكن إجراء اختبارات مرض التلاسيميا للجنين قبل الولادة، ويكون إما بأخذ عينة من مشيمة الأم الحامل، أو بتحليل عينة من السائل المحيط بالجنين.
ويتم الخضوع لعلاج مرض التلاسيميا في الحالات المتوسطة والشديدة، ويتضمن العلاج ما يلي:
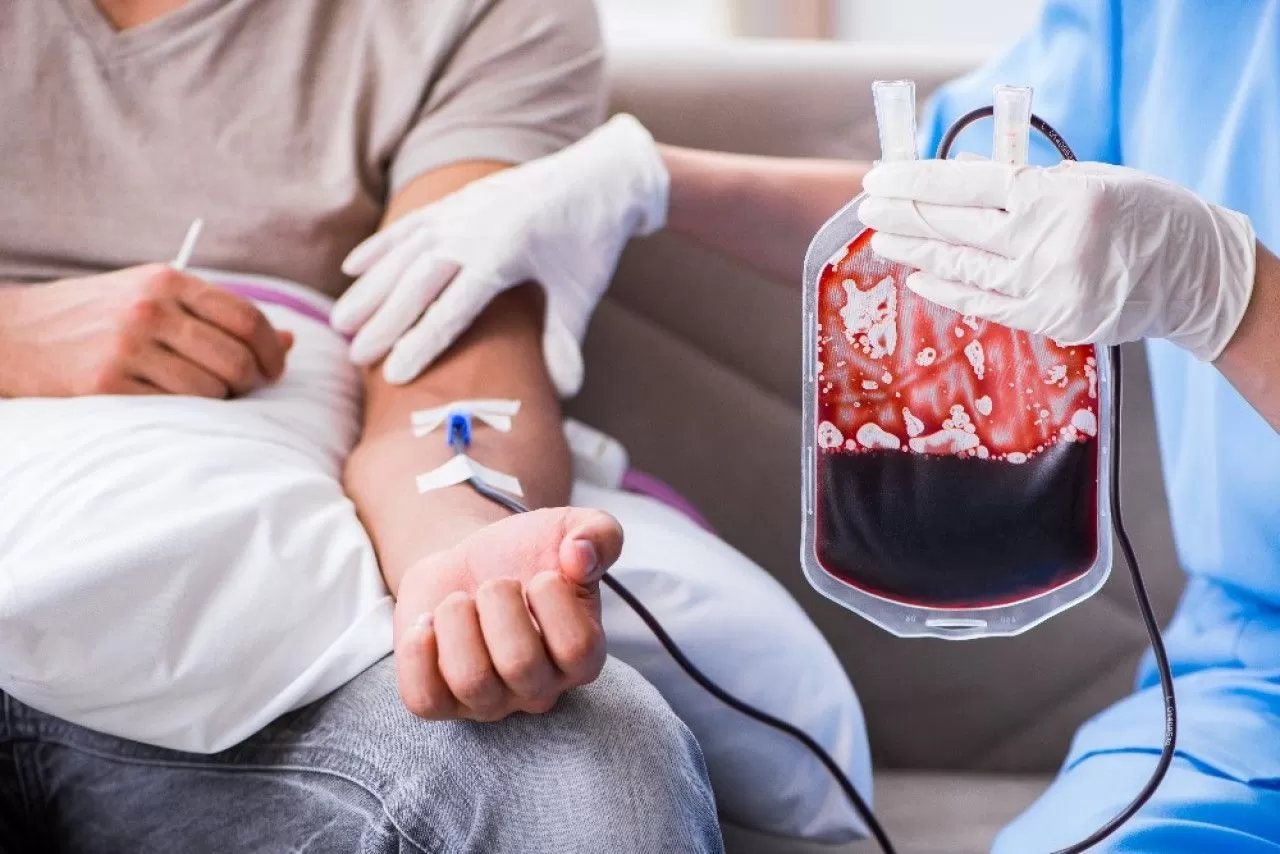
_ عمليات نقل الدم، وتتم كل بضعة أسابيع بشكل منتظم، ويُلجأ إليها في الحالات الشديدة، ولكن هذه العمليات قد تتسبب بتراكم عنصر الحديد في الدم، وبالتالي إلحاق الضرر بالقلب أو الكبد أو أعضاء أخرى من الجسم.
_ العلاج بالاستخلاب، للتخلص من نسبة الحديد الزائد في الدم، نتيجة عمليات نقل الدم، ويمكن أن يتراكم الحديد لدى مرضى التلاسيميا حتى لو لم يخضعوا لنقل الدم، ولتخليص الدم من فرط الحديد توصف أدوية يتم تناولها عن طريق الفم، مثل ديفيراسيروكس، أو ديفيريبرون، أو عن طريق الحقن مثل ديفيروكسامين.
_ زراعة الخلايا الجذعية، أو ما يسمى بزراعة نخاع العظم، وذلك بتلقي دفعات من الخلايا الجذعية من متبرع متوافق كأحد الإخوة، ويتم اللجوء لهذا العلاج عند الأطفال المصابون بالتلاسيميا الشديدة، ولا يحتاجون لعمليات نقل الدم أو أدوية السيطرة على الحديد الزائد بعد هذا الإجراء.
فقر الدم المنجلي مرض وراثي شائع في الشرق الأوسط، يصيب الأطفال منذ الولادة، وينتج بسبب عدم توافر خلايا الدم الحمراء بشكل كافٍ لنقل الأوكسجين إلى خلايا الجسم. وعندما يصاب الشخص بمرض التلاسيميا يتوقف الجسم عن إنتاج الكريات الحمراء، أما في حالة فقر الدم المنجلي، فيتم إفراز الكريات الحمراء بشكل عير طبيعي.
وتظهر أعراض فقر الدم المنجلي بعد ستة أشهر من الإصابة بالمرض، وتختلف بين فرد وآخر، وتتشابه أعراضه مع بعض أعراض مرض التلاسيميا، وأبرزها الشعور بالتعب الشديد، وصداع، وتورم الأطراف، مع تضخم في الطحال، بالإضافة لفقر الدم، وانسداد الأوعية الدموية، مع التهابات جرثومية حادة.
أخبار قد تهمك:





18 مايو 2024
18 مايو 2024
